
Variabilita prejavov osteogenesis imperfecta v závislosti na pohlaví: kazuistika
Variability of osteogenesis imperfecta manifestations depending on gender: case report
Osteogenesis imperfecta is a rare bone dysplasia with both autosomal dominant and autosomal recessive forms of inheritance. Our case presents two siblings of opposite sex with this disorder whose clinical picture and manifestation of the disease in spite of close relationship are different. The case also points out the results of five year treatment of these individuals with regards to differences between oral and intravenous use of bisphosphonates.
Keywords:
bisphosphonates – bone dysplasia – osteogenesis imperfecta
Autoři:
Štěpán Jakub; Kužma Martin; Killinger Zdenko; Payer Juraj
Působiště autorů:
V. interná klinika LF UK a UNB, Nemocnica Ružinov, Bratislava
Vyšlo v časopise:
Clinical Osteology 2019; 24(1): 36-41
Kategorie:
Kazuistika
Souhrn
Osteogenesis imperfecta je vzácna kostná dysplázia s autozomálne dominantným aj autozomálne recesívnym typom dedičnosti. Naša kazuistika prezentuje prípad dvoch súrodencov opačného pohlavia postihnutých touto chorobou, u ktorých sa napriek úzkej príbuznosti fenotypový prejav ochorenia odlišuje. Kazuistika tiež prezentuje výsledky 5-ročnej liečby postihnutých jedincov s ohľadom na rozdiely pri perorálnom a intravenóznom podávaní bisfosfonátov.
Klíčová slova:
bisfosfonáty – kostná dysplázia – osteogenesis imperfecta
Úvod
Osteogenesis imperfecta je spoločné označenie pre heterogénnu skupinu chorôb spojiva charakterizovanú celoživotným sklonom ku opakovaným zlomeninám a nízkou kostnou hmotou. Pri tomto ochorení dochádza k redukcii kortikálnej aj trabekulárnej zložky kosti [1]. Incidencia ochorenia je 1 prípad na 20 000 (živo) narodených detí [2].
Klasifikácia
Pôvodná klasifikácia osteogenesis imperfecta, ktorú publikovali Sillence et al v roku 1978 [3] rozdeľovala chorobu do 4 skupín podľa klinického obrazu a typu dedičnosti. V roku 2004 rozšírili túto klasifikáciu autori Glorieux a Rauch [1]. Za posledných 10 rokov došlo k výraznému pokroku v oblasti poznatkov o etiológii a patogenéze ochorenia. Vďaka pokrokom v genetike a molekulárnej biológii boli objavené nové autosomálne dominantné i autosomálne recesívne formy choroby, ktorej príčinou je defekt v niektorom z procesov pri zložitej syntéze molekuly kolagénu, prípadne nefunkčná mineralizácia kolagénu, či porucha pri diferenciácii osteoblastov. V súčasnosti existuje 16 typov osteogenesis imperfecta, ktoré sa líšia genetickou mutáciou [2]. Týchto 16 typov je začlenených podľa prevažujúceho klinického obrazu do klinickej (syndromologickej) klasifikácie, ktorá vychádza z pôvodnej klasifikácie podľa Sillence et al [3], ktorú upravili Glorieux a Rauch [1] a ktorá rozlišuje 5 skupín osteogenesis imperfecta I-V. V literatúre je popisovaná aj molekulárno-genetická klasifikácia, ktorá triedi jednotlivé kauzálne gény do skupín podľa ich funkcie pri syntéze kolagénu a tvorbe kosti [4].
Príznaky
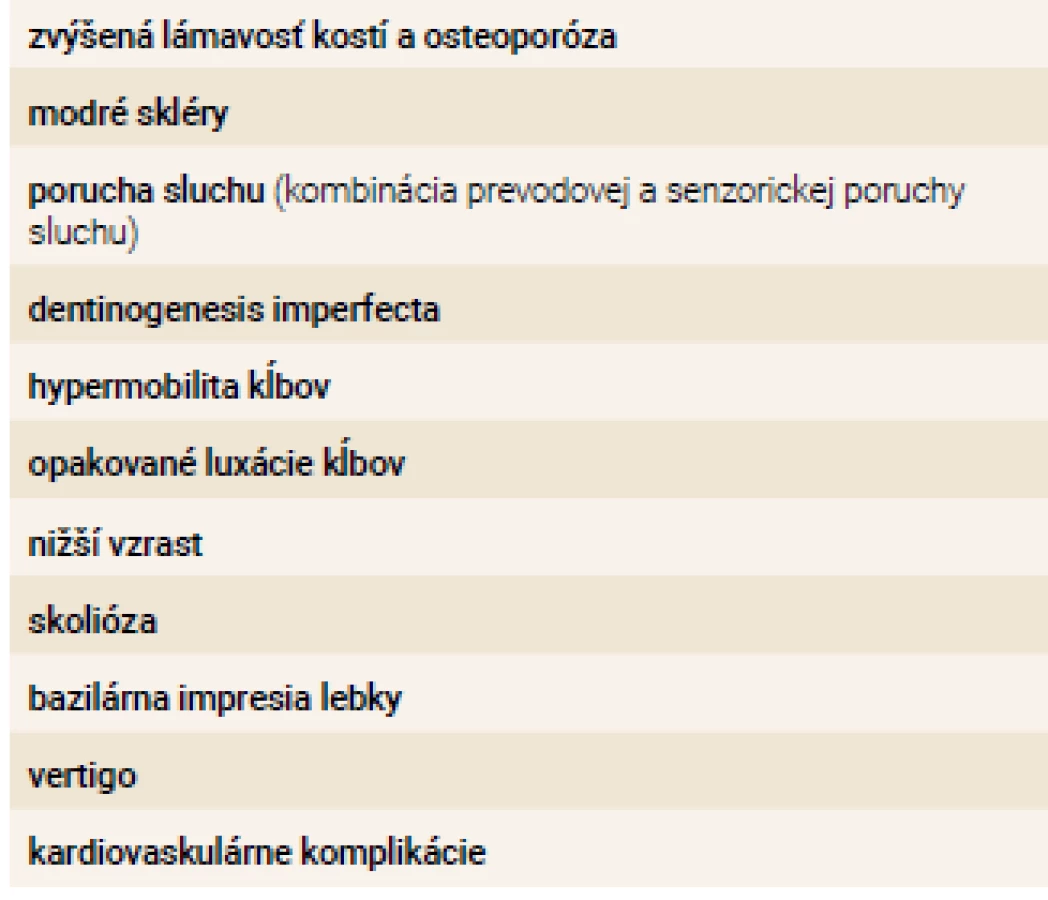
Osteogenesis imperfecta (OI) má variabilný fenotypový prejav (prejavy ochorenia, tab. 1). To znamená, že u rôznych typov ochorenia sú príznaky zastúpené v rôznej miere a ich závažnosť kolíše podľa kauzálneho génu. Medzi charakteristické príznaky ochorenia patrí zvýšená lámavosť kostí behom života, modré skléry, porucha tvorby dentície, zvýšená mobilita kĺbov, zvýšená voľnosť väzov a poruchy sluchu. Pri niektorých formách ochorenia sa tiež vyskytujú kardiovaskulárne komplikácie (ako napr. dilatácia bulbu aorty a aortálna insuficiencia). Sekundárne táto choroba vedie k nižšiemu vzrastu postihnutých jedincov a ku skolióze.
Diagnostika a diferenciálna diagnostika
V diagnostike je dôležitá pozitívna rodinná anamnéza ochorenia spolu s údajom o opakovaných často nízkotraumatických zlomeninách. Chorobu potvrdí genetické vyšetrenie s presným určením kauzálneho génu.
V diferenciálnej diagnostike je potrebné myslieť na rachitídu a syndróm týraného dieťaťa (Child Abuse and Neglect – CAN), ktorý je niekedy veľmi ťažko odlíšiteľný. K odlíšeniu CAN od OI okrem starostlivo odobratej anamnézy napomôže aj údaj o mechanizme vzniku úrazu, radiologický nález zlomenín typickejších pre CAN (symetrické zlomeniny rebier, zlomeniny metafýz) alebo extraskeletálne poranenia, napr. bilaterálne retinálne hemoragie, ktoré sú prítomné pri CAN, a naopak nevyskytujú sa u ľudí s OI [5].
Liečba
Liečba OI sa opiera o farmakoterapiu bisfosfonátmi a suplementáciu vitamínom D a vápnikom. Symptomaticky tlmíme bolesti. Operačná liečba má miesto pri rekonštrukcii zložitých zlomenín, umožňuje rýchlejšiu mobilizáciu a zmiernenie kostnej straty pri imobilizácii pacienta. Rehabilitácia slúži k obnoveniu fyzickej kondície.
Kazuistika
Jedná sa o dvoch súrodencov, 38-ročný pacient a jeho sestra, 43-ročná pacientka s pozitívnou rodinnou anamnézou osteogenesis imperfecta diagnostikovanou u ich otca, otcovej sestry a otcovej matky. Od septembra 2013 sú dispenzarizovaní v endokrinologickej ambulancii V. internej kliniky LF UK a UNB v Bratislave.
Pacient (brat)
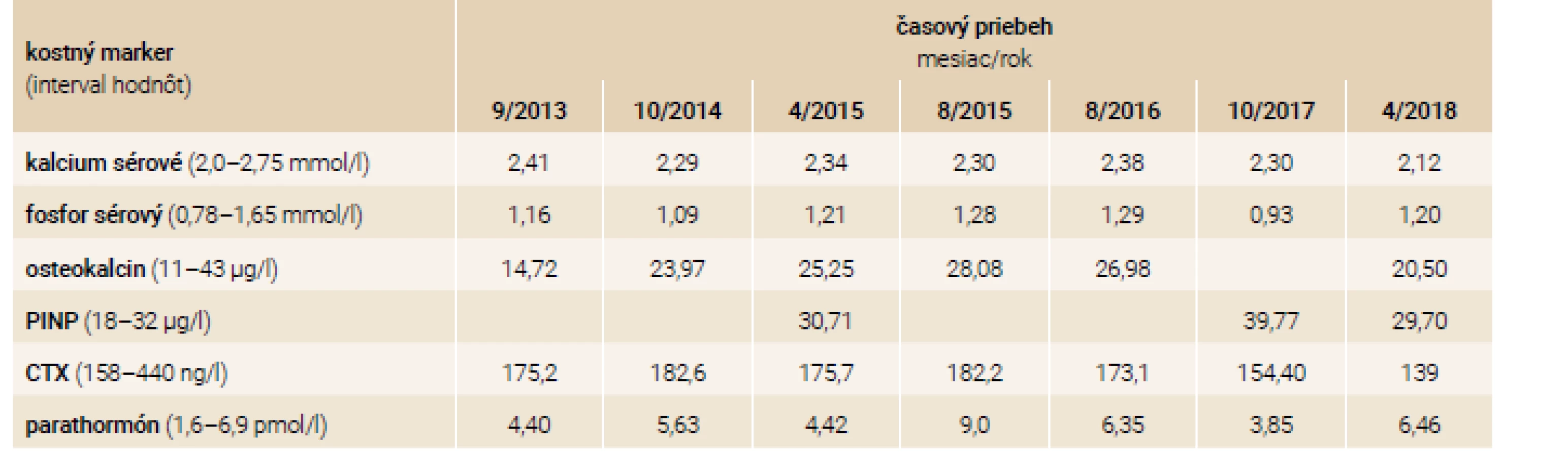
V osobnej anamnéze 38-ročného pacienta, brata 43-ročnej pacientky s rovnakou anamnézou, je približne 50 prevažne nízkotraumatických fraktúr za život, opakované hospitalizácie, z ktorých jedna bola komplikovaná rozvojom septického stavu a nutnosťou prechodnej analgosedácie na klinike anesteziológie a intenzívnej medicíny. Ďalej v osobnej anamnéze nachádzame rozvoj depresívnej poruchy. Pacient bol pred príchodom do endokrinologickej ambulancie liečený kalcitoninom (Miacalcic), alendronátom (Fosamax), jednorazovo mu bol podaný intravenózne pamidronát 60 mg/týždeň počas 3 po sebe idúcich týždňov (po odznení vážneho stavu pri zlomeninách skeletu komplikovaných sepsou). Pred rokom 2013 ešte užíval rizedronát (Risendros 35 mg perorálne 1-krát týždenne prípadne Actonel 75 mg perorálne 2-krát do mesiaca). Od roku 2014 dostáva pravidelne na jeseň zoledronát intravenózne v dávke 5 mg každých 12 mesiacov. Klinickým vyšetrením v endokrinologickej ambulancii boli zistené typické črty ochorenia: modré skléry, deformity a skrátenia dolných končatín po opakovaných fraktúrach, nižšia výška oproti bežnej populácii. Pacient netrpí poruchou sluchu a pohybuje sa pomocou bariel. Laboratórne nálezy v sledovanom období sú zhrnuté v tab. 2.
Pacientka (sestra)
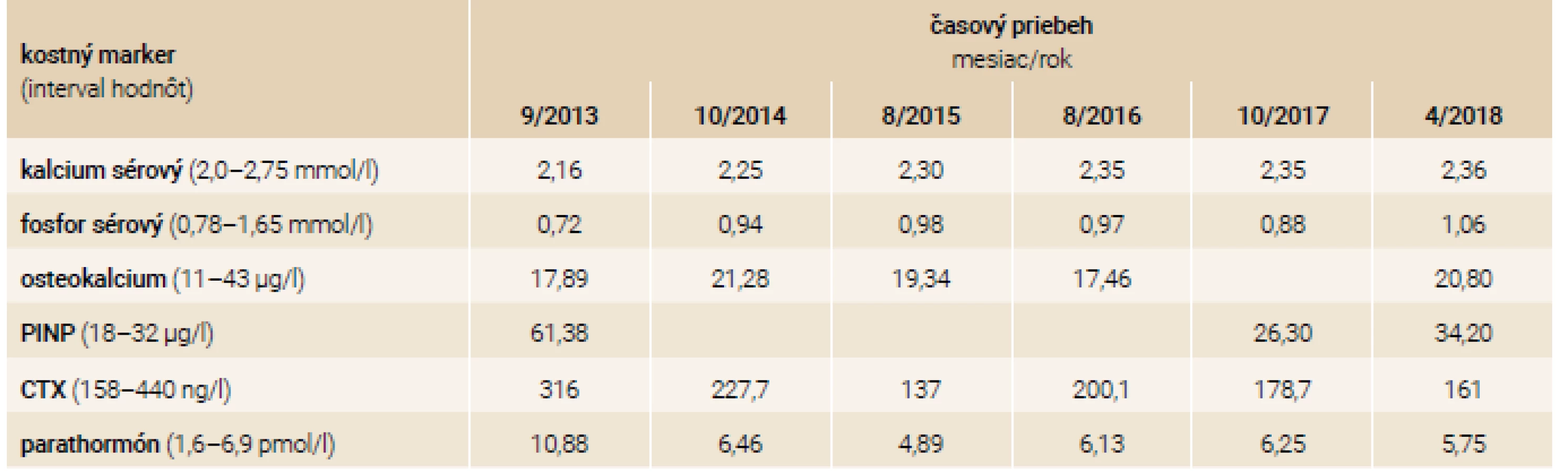
U 43-ročnej pacientky, sestry 38-ročného pacienta, je priebeh ochorenia odlišný. Do 30 rokov u nej prevládajú najmä opakované luxácie ramenných kĺbov. Po 30. roku života sa aj u nej objavujú opakované zlomeniny s frekvenciou asi 4 fraktúr/rok, pričom 1-krát utrpela závažnú kraniotraumu s mnohonásobnými zlomeninami skeletu. V osobnej anamnéze nachádzame podobne rozvoj schizoidnej poruchy osobnosti. Pacientka pred príchodom do endokrinologickej ambulancie užívala rizedronát perorálne (Risendros 35 mg perorálne 1-krát týždenne prípadne Actonel 75 mg perorálne 2-krát do mesiaca), od roku 2014 je liečená intravenózne podávaným zoledronátom v dávke 5 mg každých 12 mesiacov. Pri klinickom vyšetrení v našej ambulancii u nej nachádzame, podobne ako u brata, modré skléry, deformity a skrátenia dolných končatín po opakovaných zlomeninách, nižší vzrast oproti populačnému priemeru. Pacientka podobne ako brat nemá poruchu sluchu. Po zahájení liečby zoledronátom dochádza u pacientky až na jednu výnimku k vymiznutiu zlomenín v sledovanom období. Po 4 rokoch liečby intravenózne podávaný zoledronát nahradzujeme na žiadosť zubného lekára perorálne podávaným rizedronátom z dôvodu obavy pred osteonekrózou čeľuste pri extrakcii zubu. Približne 3 mesiace po zahájení terapie rizedronátom dochádza u pacientky k 2 nízkotraumatickým zlomeninám: zlomenine pravej ulny a abrupcii trochanter major pravej stehennej kosti. Laboratórne nálezy pacientky v sledovanom období sú zhrnuté v tab. 3.
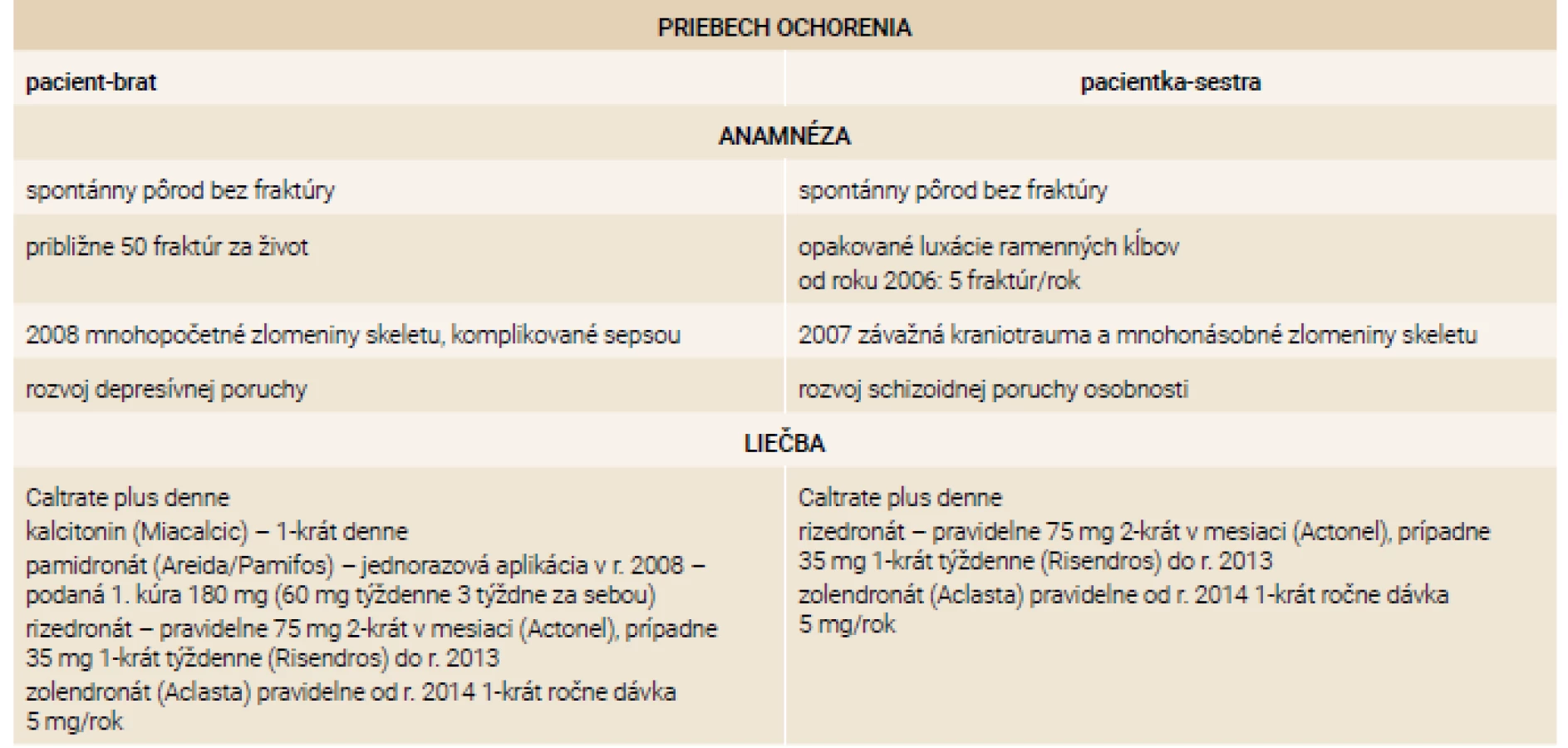
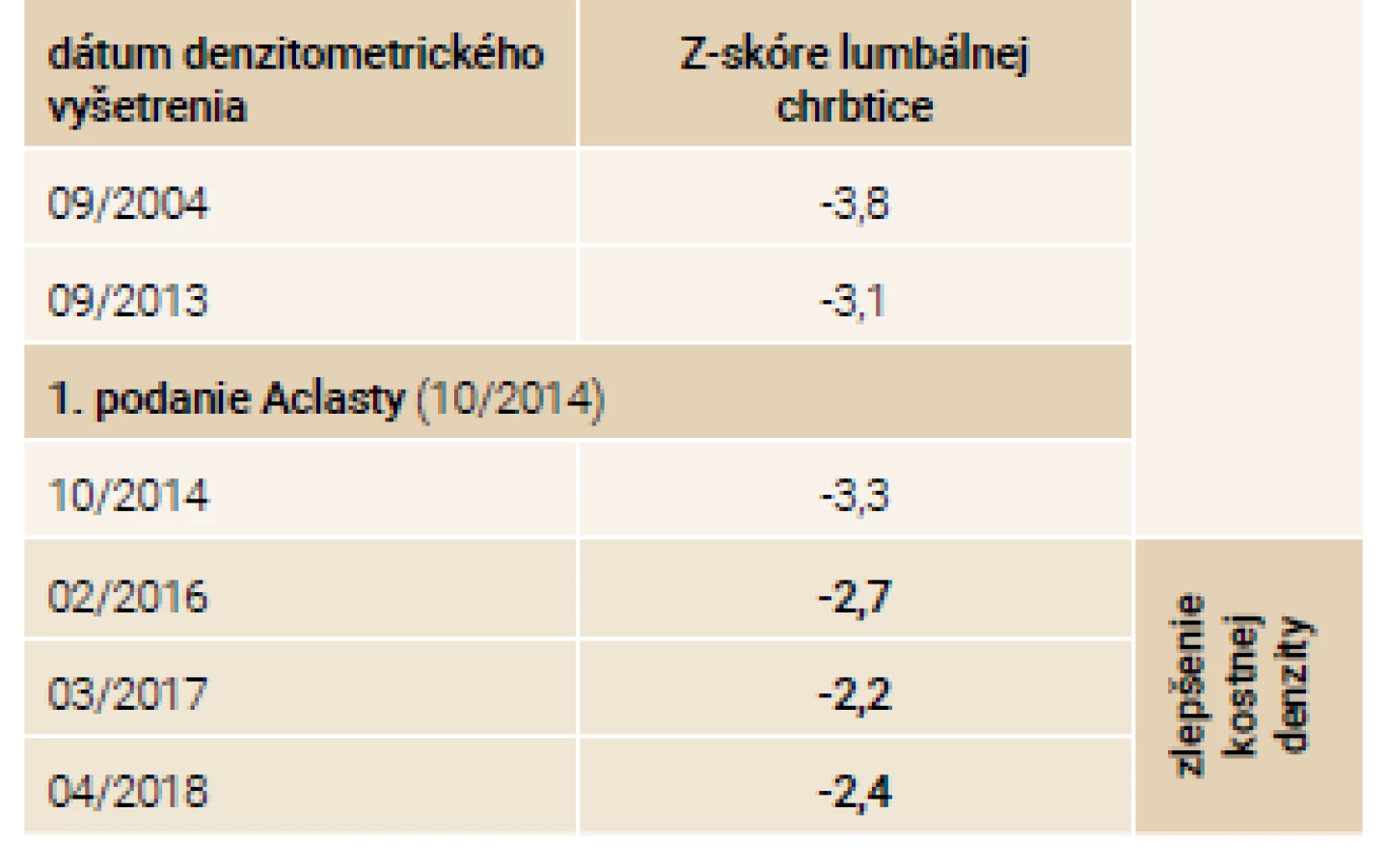
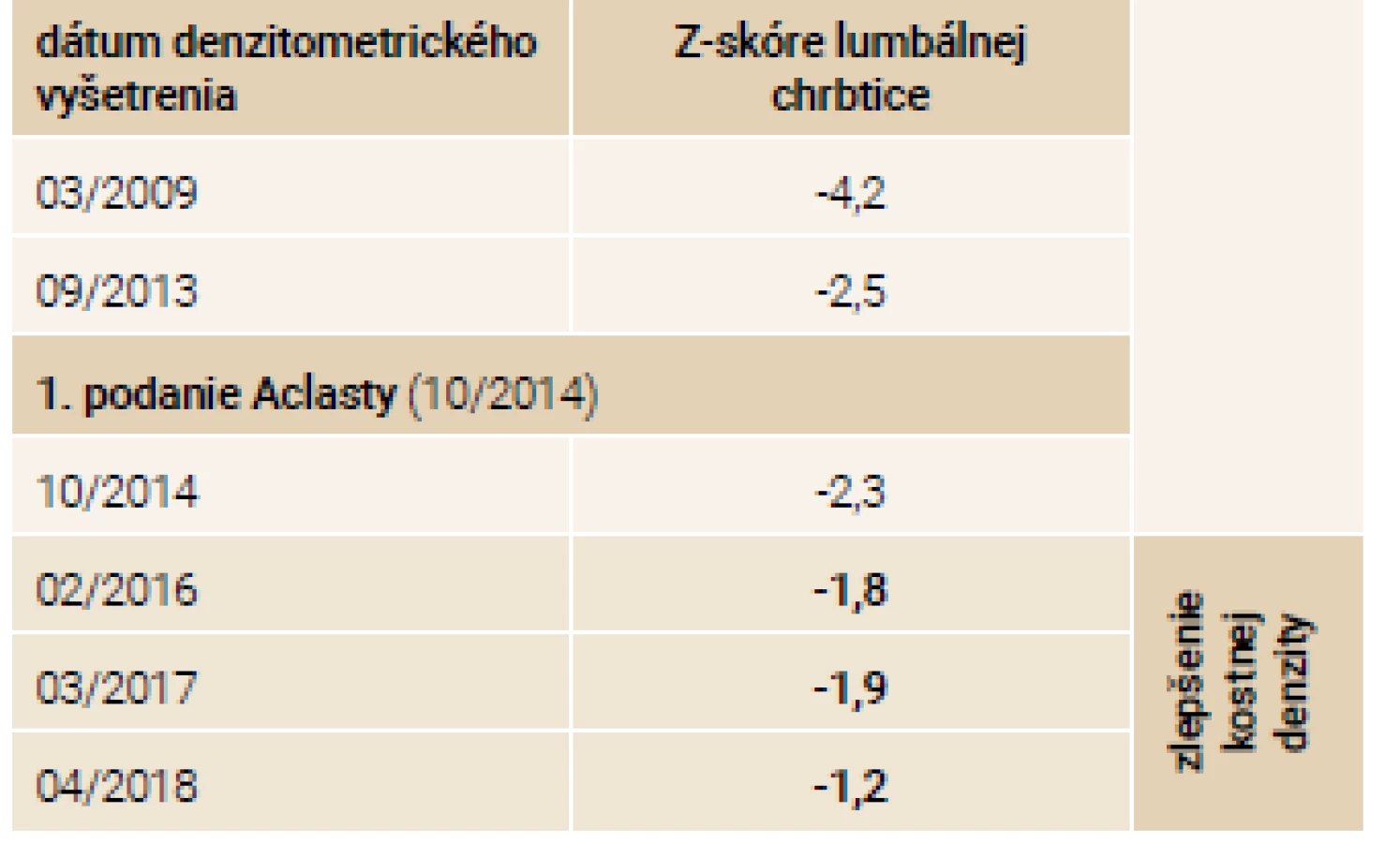
Priebeh ochorenia u súrodencov zobrazuje tab. 4.
Doplnené genetické vyšetrenie oboch pacientov preukazuje kauzálnu mutáciu c769C>T(p.Gly257Arg) v géne COL1A1. Stanovená diagnóza je osteogenesis imperfecta typ 1, autosomálne dominantne dedičná s variabilnou klinickou expresivitou (obr).


Liečba v endokrinologickej ambulancii
Na jeseň v roku 2014 zahajujeme terapiu intravenózne podávaným zoledronátom v dávke 5 mg 1-krát ročne. Počas sledovaného obdobia 5 rokov od 9/2013 do 12/2018 dochádza ku zlepšeniu klinického stavu oboch pacientov. Subjektívne udávajú zlepšenie chronických kostných bolestí a zvýšenie mobility. Aj napriek pádom sa v tomto období objavila len 1 zlomenina pately u sestry v roku 2014. Doplnené denzitometrické vyšetrenia lumbálnej chrbtice odrážajú toto klinické zlepšenie v podobe nárastu kostnej hmoty o približne 10 % u oboch súrodencov tab. 5 a tab. 6.
Diskusia
Osteogenesis imperfecta je vzácne metabolické ochorenie kostí, ktorého incidencia sa pohybuje na úrovni 1 : 20 000 narodených detí, pričom objavenie nových kauzálnych génov, hlavne fenotypovo klinicky menej výrazných túto incidenciu môže ešte zvyšovať. Počet rodín s touto chorobou na Slovensku nie je presne známy. V posledných rokoch viedlo objavenie nových foriem choroby opätovne ku vzbudeniu záujmu vedeckých tímov o túto diagnózu. Navyše pochopenie patofyziológie a patogenézy tejto choroby prispieva spätne k poznaniu fyziologického rozvoja kosti a spojivových tkanív. Asi 90 % prípadov choroby súvisí s autosomálne dominantne dedičnou poruchou tvorby kolagénu v géne COL1A1, resp. COL1A2 [4]. Vzniká buď normálny kolagén, ale v nedostatočnom množstve, prípadne defektný kolagén v normálnom množstve. To je aj prípad v našej kazuistike, v ktorej mali pacienti identifikovanú mutáciu v géne COL1A1. Naopak 10 % ochorenia predstavujú vzácnejšie formy s autosomálne dominantnou i recesívnou formou dedičnosti. Jedná sa napríklad o gény kódujúce enzymatické komplexy, ktorých úlohou je posttranslačná modifikácia novo vzniknutých alfa-reťazcov kolagénu. Iné kauzálne gény produkujú proteíny účastniace sa cross-linkingu molekúl tropokolagénu vo fibrilách. Fibrily sú následne stabilnejšie a odolnejšie v ťahu [4]. Pre diagnostiku ochorenia je dôležitá rodinná anamnéza a klinický obraz opakovaných nízkotraumatických zlomenín behom života. V našej kazuistike boli prítomné obidva tieto predpoklady. V liečbe OI používame metódy rehabilitácie, chirurgickú liečbu zlomenín, z farmák najmä bisfosfonáty. Podľa súčasnej evidencie neexistuje jednoznačný postup, ktorý z bisfosfonátov je najvhodnejší. Existuje málo štúdií porovnávajúcich účinok bisfosfonátov s placebom na zvýšenie kostnej hmoty a zníženie množstva zlomenín. Tieto štúdie navyše obsahujú vzhľadom ku vzácnosti ochorenia malé súbory pacientov. Dokonca metaanalýza dvoch Cochrane reviews a jednej placebom kontrolovanej štúdie nepodporila štatisticky významný efekt bisfosfonátov na množstvo zlomenín u ľudí s OI [7]. To je v rozpore s našou skúsenosťou a obecne bisfosfonáty stále zostávajú hlavnou farmakologickou liečbou ochorenia [6]. Stále však nie je jasné, ktorý bisfosfonát použiť, a či má cesta podania lieku efekt na morbiditu pacienta, prípadne na subjektívne pociťovanie bolesti. Vyskočil et al potvrdil pozitívny vplyv perorálne podávaného alendronátu na kostnú denzitu, subjektívne vnímanie chronickej bolesti, aj na redukciu množstva fraktúr u skúmanej vzorky 30 pediatrických pacientov počas 3 rokov sledovania [8]. V našej kazuistike boli obaja súrodenci liečení perorálne rizedronátom v dávke 35 mg týždenne (alebo tiež Actonel 75 mg 2-krát mesačne perorálne) do roku 2013, avšak bez dostatočného efektu na BMD a incidenciu zlomenín. Po príchode pacientov do nášho osteocentra sme začali terapiu zoledronátom v dávke 5 mg každých 12 mesiacov, ktorá viedla k redukcii zlomenín oboch pacientov behom prvých 4 rokov aplikácie lieku aj napriek tomu, že mali rozdielnu klinickú manifestáciu choroby najmä pred 30. rokom veku.
Účinnosť zoledronátu je v našej kazuistike podporená aj faktom, že pri jeho prechodnom vysadení u našej pacientky (nebola podaná dávka na jeseň v r. 2018) a nahradení menej potentným perorálnym rizedronátom na žiadosť zubného lekára pre plánovanú extrakciu zubov a riziko vzniku osteonekrózy čeľuste došlo u pacientky v zime v roku 2019 ku nízko traumatickej zlomenine pravej ulny a abrupcii trochanter major pravej stehennej kosti. Efekt zoledronátu odráža aj vzostup Z-skóre v oblasti lumbálnej chrbtice. V oblasti krčka stehennej kosti nebolo možné realizovať merania, pretože po zlomeninách sa v tejto oblasti vyskytovali osteoproduktívne zmeny.
Rozdielny fenotypový prejav ochorenia v jednej rodine u súrodencov opačného pohlavia nie je zatiaľ objasnený. Štúdie na humánnych modeloch zaoberajúce sa fenotypovým prejavom OI v závislosti na pohlaví neexistujú. Modelovaná štúdia s myšami však ukazuje, že mužské pohlavie má väčšiu biomechanickú odolnosť kosti, ktorá je dôsledkom rozdielnej štruktúry a geometrie pri raste kosti [9].

Záver
Osteogenesis imperfecta je raritným metabolickým ochorením kostí, ktoré sa prejavuje heterogénnym klinickým obrazom nielen v rámci jednotlivých typov, ale aj v tom istom type ochorenia , čo potvrdzuje uvedená kazuistika dvoch súrodencov s odlišnými klinickými prejavmi ochorenia. K diagnostikovaniu choroby dochádza často v detskom veku na základe opakovaných nízkotraumatických zlomenín u jedinca. Prevalencia ochorenia môže mierne narastať v súvislosti s rozvojom poznatkov najmä v oblasti genetiky tejto choroby, pretože sú spoznávané aj jej miernejšie formy.
Vo farmakoterapii choroby je liekom voľby antiresorpčná liečba bisfosfonátmi. Ostatné modality liečby zahŕňajú rehabilitačnú a chirurgickú liečbu, ktorých cieľom je skrátiť čas imobilizácie a predchádzať tak kostným stratám pri inaktivite.
MUDr. Jakub Štěpán
Doručené do redakcie 28. 5. 2019
Prijaté po recenzii 28. 6. 2019
Zdroje
- Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 2014; 164A(6): 1470–1481. Dostupné z DOI: <http://dx.doi.org/10.1002/ajmg.a.36545>. Erratum in Am J Med Genet A 2015; 167A(5): 11 78.
- Beary JF, Chines AA, Firth HV. Osteogenesis imperfecta: Clinical features and diagnosis. Dostupné z WWW: <https://www.uptodate.com/contents/osteogenesis-imperfecta-clinical-features-and-diagnosis>.
- Sillence DO, Rimoin DL. Classification of osteogenesis imperfect. Lancet 1978; 13 (1/8072): 1041–1042.
- Forlino A, Marini JC. Osteogenesis imperfecta. Lancet 2016; 387(10028): 1657–1671. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(15)00728-X>.
- Pereira EM. Clinical perspectives on osteogenesis imperfecta versus non-accidental injury. Am J Med Genet C Semin Med Genet 2015; 169(4): 302–306. Dostupné z DOI: <http://dx.doi.org/10.1002/ajmg.c.31463>.
- Beary JF, Chines AA, Firth HV. Osteogenesis imperfecta: Management and prognosis. Dostupné z WWW: <https://www.uptodate.com/contents/osteogenesis-imperfecta-management-and-prognosis>.
- Dwan K, Phillipi CA, Steiner RD et al. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev 2014; 7: CD005088. Dostupné z DOI: <http://dx.doi.org/10.1002/14651858.CD005088.pub3>.
- Vyskocil V, Pikner R, Kutílek S. Effect of alendronate therapy in children with osteogenesis imperfecta. Joint Bone Spine 2005; 72(5): 416–423. Dostupné z DOI: <https://doi.org/10.1016/j.jbspin.2004.09.005>.
- Yao X, Carleton SM, Kettle AD et al. Gender-dependence of bone structure and properties in adult osteogenesis imperfecta murine model. Ann Biomed Eng 2013; 41(6): 1139–1149. Dostupné z DOI: <https://doi.org/10.1007/s10439–013–0793–7>.
Štítky
Biochemie Dětská gynekologie Dětská radiologie Dětská revmatologie Endokrinologie Gynekologie a porodnictví Interní lékařství Ortopedie Praktické lékařství pro dospělé Radiodiagnostika Rehabilitační a fyzikální medicína Revmatologie Traumatologie OsteologieČlánek vyšel v časopise
Clinical Osteology

2019 Číslo 1
Nejčtenější v tomto čísle
- Sarkopenie: definice a diagnostika nové nemoci
- Vliv vitaminu K na muskuloskeletální zdraví u postmenopauzálních žen
- Variabilita prejavov osteogenesis imperfecta v závislosti na pohlaví: kazuistika
- Efekt dlhodobej liečby rastovým hormónom na kostnú hustotu a kvalitu kosti